How To:
1. How to view a molecule
(a) click the "File/Open" menu, check the radio button "XYZ", and locate the molstart directory to open the buckminsterfullerine.xyz file.
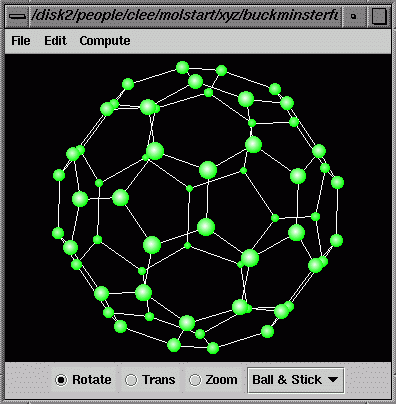
(b) Click the "File/Open"
menu, check the PDB radio button and locate the molstart
directory to open pkc.pdb file. Select "Stick" or "Ball &
Stick" to toggle chemical bonds in different styles. The current version
of MolStart displays only the "ATOM"
part of the pdb file, "HETATM" is not included in the molecular
structure. MolStart has been tested to read pdb file with more than 15,000
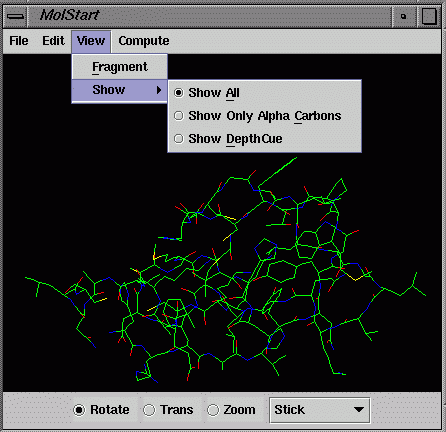
atoms. The default display only shows Alpha Carbons if the total
number of atoms is greater than 7,000. Click on the menu "View/Show"
and select "Show All" or other options to toggle between
different display styles. Select the "Show Only Alpha Carbons"
to Rotate/Trans/Zoom for a large molecular system to get quick response.
2. How to view bond distance, bond angle
and dihedral angle
Select the "Edit" menu and
select "Measurement/Modify" to pop up the "Measurement" window. Click
the "Bond" button and select two atoms in the Molecular
Structure window to view bond distance between the two
selected atoms
. Click the "Angle" button and select
three atoms to view the bond angle. Click "Dihedral" and select four
atoms to view the dihedral angle.
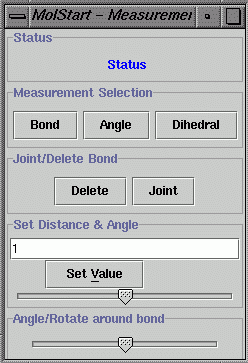
3. How to set a new bond distance
Click the "Bond" button and select two atoms
on the Molecular Panel. Specify a value in "Set Distance & Angle" box
or drag the sliding bar to change the bond distance visually. Click on the
"Set Value" button to set the bond length between the two atoms to
the specified value. Please note that the value is committed only
when the Molecular Structure Panel is clicked afterwards when using the sliding
bar. The first selected atom with other connected atoms will move to the
new distance.
4. How to set bond angle
Click on the "Angle" button and select three atoms on
the Molecular Panel. Specify an "Angle Value" and click on the "Set Value"
button to set the angle to the specified value or drag the "Angle/Rotate around bond"
bar to change the angle visually. Please note that the value is committed only when the
Molecular Structure Panel is clicked afterwards if using the sliding bar. The first selected
atom with other connected atoms will be set to the new value.
5. How to rotate atom(s) about a bond
Click on the "Bond" button and drag
the slider in the "Angle/Rotate around bond" to rotate the first selected atom with other connected atoms to a
new angle. Please note that the value is committed only when
the Molecular Structure Panel is clicked afterwards.
6. How to save new coordinates to a file
Click on the "File/Save" menu selection to write to
a new file, or select an old file name to overwrite it.
7. How to add
atom(s)
Click on the "Edit/Periodic Table" menu item.
An "Atom Selection" window will popup.
| Simple rules: |
|
(a) Adding atoms without valence structure: Pick an atom and click the "Keep Cursor Loaded" button. Click on the Molecular Structure Panel to add atoms.
| Two ways to add atom: |
|
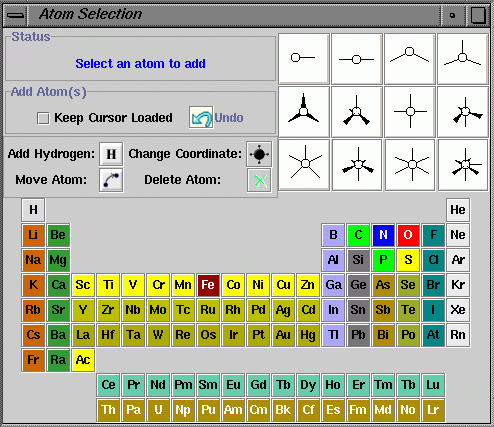
(b) Adding atom with valence structure: First select an atom from periodic table and click on the valence bond picture to select the valence associated with the atom. Then click on the Molecular Structure Panel to add atoms. There are pink color balls with red "bond" connections to the atom. The pink color balls are the indicator of the valence points. To add more atoms, select another atom and valence structure and click on the valence pink ball you wish to replace. Before joining chemical bond for two atoms, the valence pink balls that are treated like "Atom" can be deleted using "Delete Atom" button.
8. How to change valence coordinates of
an atom
Click on the desired "Valence"
icon, and click the "Change Coordinates" button and then click
on the atom in the Molecular Structure Panel to change the valence
structure.
9. How to move an atom
Select the "Edit/Periodic
Table" menu item. Click the "Move Atom" button on the "Periodic Table"
window, select an atom on the Molecular Structure Panel and drag to new position. Click anywhere on
the Molecular Structure Panel but not on any atom to unload the "Move"
function.
10. How to delete an atom
Click on the "Edit/Periodic Table" menu item. Click the "Delete
Atom" button on the popup window, select an atom on the Molecular
Structure Panel to delete the atom. Click anywhere on the Molecular Structure Panel
but not on any atom to unload the "Delete" function.
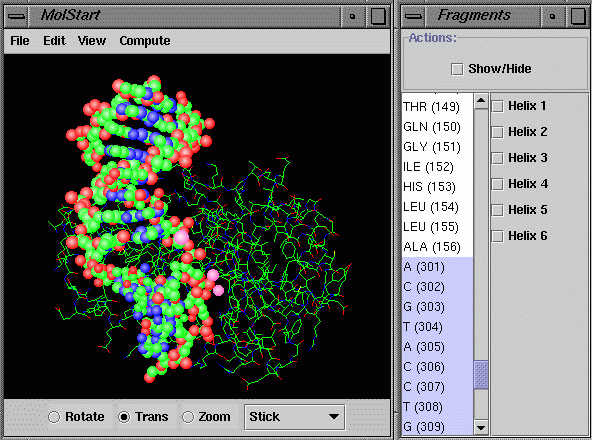
11. How to view selected amino acids or
nucleotides in the protein/DNA in PDB format
Click on
the menu "File/Open" to load a *.pdb file into MolStart molecular
structure panel. Click on the menu “Biopolymer/Residue” to pop up a “Residues”
window. Amino acids or nucleotides with the sequence number are displayed
in the list. Click on the amino acid or nucleotide shown in the list to
change the Stick to Ball&Stick for the atoms in the selected amino
acids or nucleotides. Use the “Shift” key to select a block of residues
and use the “Control” key to add individual residue selections. Click on the
"Show/Hide" checkbox to hide or show the selected residues. Click the
Helix check box to display selected helix sturcture(s).
12. How to view and edit the molecular structure
in a subset region when editing a big molecule.
1. Click the "View/Subset" menu
to popup a “Subset” window to enter "Center of Subset" and "Radius of
Subset."
2. Click an atom in the molecular structure to
define "Center of Subset" or simply just type in the sequence number of
the atom in the displayed molecular structure.
3. Click the "Execute" button to
define a "Subset". The atoms in the subset are displayed in "Ball & Stick".
4. Click the menu “View/Center the molecule” to put the subset in the
center of the molecular structure panel, so the "Rotate/Trans/Zoom"
operations are readjusted to the subset.
5. Click the
“Edit/Periodic Table” to popup Periodic table for editing the subset.
6. Click the “Clear” button or close the "Subset" window to disable
the subset.
13. How to open a PDB file from another
website
In this release, we have only implemented one
Protein Data Bank website, http://www.rcsb.org/, for submitting
queries. Click the menu, “File/Open PDB from RCSB”, to popup a “Enter a PDB
ID” window for entering PDB ID that is available in the RCSB protein data bank,
for example, 1cw0. The structure of the 1cw0 protein will be displayed in
the molecular structure panel. Please note that your computer needs to
be connected to the Internet in order to send a query to the RCSB web
site.